

Free Webinar
Hear directly from pharmaceutical companies on how they are transitioning from the traditional Limulus Amebocyte Lysate (LAL) methods to cutting-edge recombinant technologies, driving sustainability and innovation.
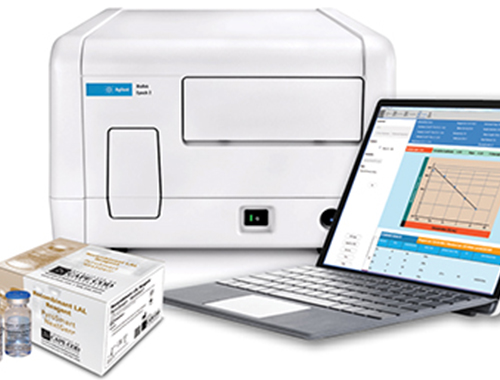
Agilent - BioTek Epoch 2
ACC’s Pyros® eXpress Software now fully compatible with the Epoch 2 Plate Reader.

Sustainability In BET!
The Future of Sustainable LAL Recombinant Cascade Reagent
Has Arrived!

More than 1 million juvenile horseshoe crabs reared and released!
Horseshoe Crab Sustainability Project focuses on supporting fisheries worldwide and ensuring the genetic diversity of the horseshoe crab.

Certificates
Most of our Certificates of Analysis, Certificates of Compliance, and Safety Data Sheets are available on-line.
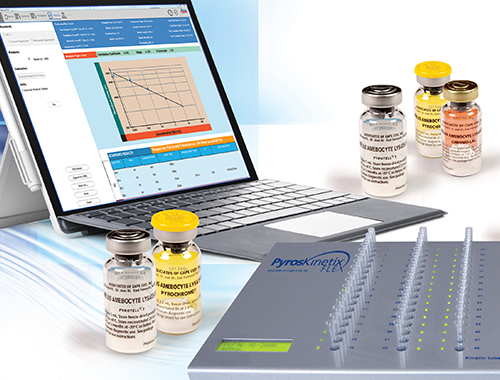
Software and Instrumentation
Endotoxin and Glucan Digital Testing and Analysis.

Pursuing Sustainability Excellence
Since 2011, Associates of Cape Cod, Inc., has had a growing commitment to taking a responsible role in making sure we do our part to ensure that we reduce the environmental footprint on our operations.

Horseshoe Crabs And The Biomedical Industry... Know The Truth
Frequently Asked Questions
Endotoxin Testing Reagents
LAL reagents are primarily used to test for endotoxins in injectable pharmaceuticals, biological products, and medical devices.

Contract Test Service
Our CTS laboratory specializes in testing for endotoxin and glucan contamination.

BET Methodology and Support Services
Through free consultation and support we will assist you to determine the method that best suits your needs.
Pyros® eXpress Software
The Next Generation of Endotoxin and Glucan Analysis Software for glass tube and plate readers from your Endotoxin experts.
Associates of Cape Cod, Inc. introduces the next generation of endotoxin and glucan detection analysis software that offers integrated solutions for your quantitative endotoxin and glucan detection testing, reporting needs, trending and data management.
Learn More